
Le manganèse comme seul traitement d’une maladie rare de la glycosylation
Dans un article publié dans la revue Translational Research, des scientifiques issus de l'Unité de Glycobiologie Structurale et Fonctionnelle (UGSF1 ) montrent qu’un simple traitement au manganèse est suffisant pour traiter une forme de maladie génétique rare de la glycosylation, processus biochimique qui permet l’ajout de sucres à des protéines ou des lipides. Ce succès a été permis par la découverte et la compréhension du rôle d’une protéine, la protéine TMEM165 spécifiquement impliqué dans cette pathologie.
- 1UGSF - CNRS/ULille
Les Anomalies Congénitales de Glycosylation (CDG), des maladies génétiques rares.
Chez l’Homme, la glycosylation est un processus enzymatique cellulaire fondamental et ubiquitaire consistant en la synthèse de structures glycaniques, appelées glycanes, portées par des lipides ou des protéines. Constituées de différents monosaccharides tels que l’acide sialique, le galactose, le mannose etc. la diversité des structures synthétisées résulte de l’activité d’enzymes localisées dans la voie de sécrétion, les glycosyltransférases. L’importance que revêt ce processus complexe est illustrée par l’existence de maladies humaines génétiques rares, les CDG (Congenital Disorders of Glycosylation ou Anomalies Congénitales de Glycosylation), résultant d’un défaut de glycosylation. Le nombre de patients CDG identifiés dans le monde ne cesse d’augmenter pour atteindre aujourd’hui plus de 170 sous-groupes différents. Malgré des efforts constants, les options thérapeutiques demeurent limitées, le plus souvent à cause d’une méconnaissance des voies métaboliques perturbées.
Un transporteur du Manganèse impliqué dans un sous-groupe de CDG
En 2012 un nouveau sous-type de CDG a été découvert, lié à des mutations dans le gène TMEM165, codant pour la protéine TMEM165, de fonction jusqu’alors inconnue. Les patients présentaient un défaut général sévère de la glycosylation, associé à un phénotype clinique spécifique de dysplasie squelettique, ostéoporose, scoliose, nanisme et déficience intellectuelle. Les scientifiques ont démontré que TMEM165 était un transporteur de manganèse localisé dans l’appareil de Golgi et essentiel à la régulation de l’homéostasie de ce dernier. Le manganèse est un cofacteur de nombreuses glycosyltransférases, qui, en s’associant à leur site catalytique, participe à leur activité. L’absence de fonctionnalité de TMEM165 entraîne une déplétion en manganèse intra-Golgien et de façon secondaire, une hypogalactosylation liée à la dysfonction des glycosyltransférases Mn-dépendantes. Un des résultats clés de ces recherches fût de démontrer que la supplémentation en Mn2+ (quelques µM seulement) suffisait à restaurer complètement les capacités de glycosylation des cellules déficientes en TMEM165, avec l’idée sous-jacente que ce concept puisse un jour être bénéfique pour des patients CDG.
Lire la suite sur le site internet de CNRS Biologie
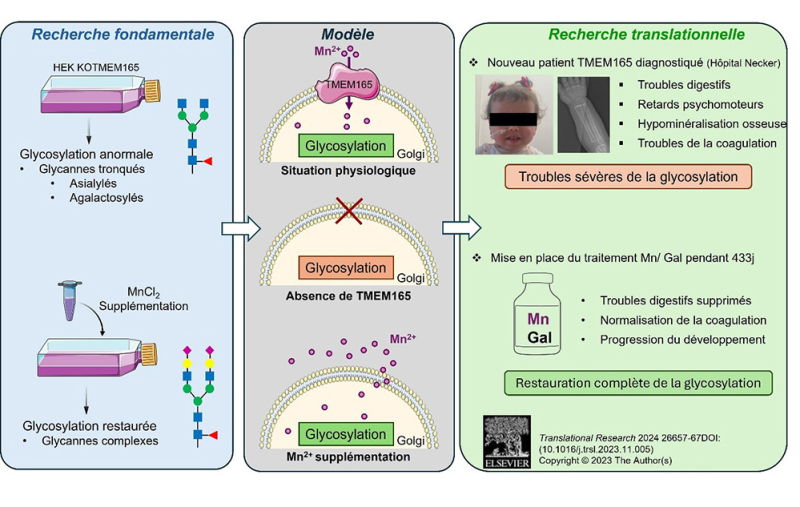
Encart in vitro : l’analyse des glycanes dans des cellules HEK KO TMEM165 révèle des structures tronquées composées de N-acétylglucosamine (carré bleu) de mannose (rond vert) et de fucose (triangle rouge), où manquent principalement les acides sialiques (losange violet) et les résidus de galactose (rond jaune). Le traitement au MnCl2 des cellules entraîne une restauration de la glycosylation, avec des structures complexes correctement galactosylées et sialylées.
Encart Modèle : en situation non pathologique, TMEM165 importe du Mn2+ (rond rose) dans l’appareil de Golgi, et la glycosylation est normale. L’absence de la protéine TMEM165 entraîne un manque de Mn2+ et subséquemment un défaut de glycosylation. Avec un traitement au manganèse, la quantité de cet ion dans le Golgi est restaurée, et permet de supprimer le défaut de glycosylation.
Encart Recherche translationnelle : La patiente diagnostiquée présente les symptômes décrits, et une glycosylation anormale. Après traitement au Manganèse (Mn) et D-Galactose (Gal), les symptômes sont améliorés, et la glycosylation est restaurée.
© Zoé Durin, Alexandre Raynor, François Fenaille, Pascale de Lonlay, Arnaud Bruneel, François Foulquier, CC BY 4.0, Servier Medical Art
Pour en savoir plus :
Efficacy of oral manganese and D-galactose therapy in a patient bearing a novel TMEM165 variant
Z.Durin, A.Raynor, F.Fenaille, P. de Lonlay, A.Bruneel, F.Foulquier
Translational Research, 25 novembre 2023. DOI : Efficacy of oral manganese and D-galactose therapy in a patient bearing a novel TMEM165 variant